
BAM Chapter 26: Detection and Quantitation of Hepatitis A Virus in Shellfish by the Polymerase Chain Reaction
Bacteriological Analytical Manual (BAM) Main Page
January 2001
Author: Biswendu B. Goswami (ret.)
For additional information, contact Michael Kulka.
Hepatitis A virus (HAV), a major cause of infectious hepatitis in humans, is a positive strand RNA virus belonging to the hepatovirus group of the picornavirus family. Primary detection of HAV in clinical or biological samples is not routinely possible at present because wild-type HAV grows very poorly in cell culture. Except for virus preparations that have been adapted for rapid growth in cell culture, HAV does not produce a detectable cytopathic effect in infected cells. For these reasons, several laboratories have attempted to develop methods based on nucleic acid hybridization or the polymerase chain reaction (PCR) for detecting HAV. The reverse transcription-PCR (RT-PCR) procedure described here for detecting HAV in shellfish can be adapted for use in clinical samples, which have a much higher viral load and which contain less extraneous material that could interfere with RT-PCR. Also described here are the synthesis and use of a competitor template RNA for determination of the number of HAV genomic RNA molecules in a sample. We emphasize, however, that the method described here does not provide information about the infectious viral load in a sample, but simply the number of RNA molecules having the HAV specific sequence.
- Equipment and materials
- DNA thermal cycler
- Agarose gel electrophoresis set-up
- Gel electrophoresis power supply
- UV box
- Liquid scintillation spectrometer
- Computer for regression analyses of [32P]nucleotide incorporation data
- Water saturated phenol (from supplier)
- 10 mM Tris-HCl (pH 8.00) - 1 mM EDTA (TE)
- TE saturated phenol:chloroform:isoamyl alcohol (24:24:1) (PCIA)
- Plasmid DNA purification kit (QIAGEN)
- Eppendorf refrigerated microcentrifuge
- Refrigerated high speed centrifuge
- Water bath
- Restriction enzymes BamHI and HindIII (see ref. 1); also EcoRI.
- SP6 RNA polymerase
- Reverse transcriptase
- Thermostable DNA polymerase
- 3 M sodium acetate (pH 5.5)
- RNAse-free DNAse
- Vanadyl ribonucleoside complex
- Forward primer 5'ATGCTATCAACATGGATTCATCTCCTGG3'
- Reverse primer 5'CACTCATGATTCTACCTGCTTCTCTAATC3'
- RNAse inhibitor, human placenta
- Oligo (dT)12-16
- Unlabeled dNTPs
- [32P]dATP or dCTP
- Plasmid pHAV6 (available from author)
-
Synthesis of competitor template RNA
The competitor template RNA is synthesized in vitro from the plasmid pHAV6 with SP6 RNA polymerase. The competitor RNA harbors a 63 base deletion in the region amplified by the PCR primer pair. This enables separation of the PCR product originating from wild-type viral RNA from the product generated from the competitor RNA in the same reaction tube. The design and construction of the plasmid pHAV6 was described in detail elsewhere (1).
For RNA synthesis, purify the plasmid by using a Qiagen plasmid kit and following the manufacturer's instructions. Linearize the plasmid by using EcoRI; synthesize RNA and purify as previously described (1).
-
Isolation of tissue RNA from hard shell clams
Virtually any method of RNA isolation that provides good quality RNA will suffice. Isolate RNA by direct homogenization of fresh or frozen tissue in 10 volumes of 50 mM sodium acetate buffer (pH 5.5)-2 mM EDTA-1% SDS, and 10 volumes of water-saturated phenol in an Omni mixer at full speed for 1 min. Shake the mixture vigorously for 10 min and centrifuge (all centrifugations are at 10,000 x g for 10 min at 4C unless otherwise stated) to separate the phases. Re-extract the upper aqueous layer with an equal volume of water-saturated phenol as described above. Adjust the aqueous phase to 0.2 M in sodium acetate and precipitate total nucleic acids with 2.5 volumes of ethanol. Collect the bulky precipitate by centrifugation. Remove contaminating DNA, tRNA, and polysaccharides by three successive washes with ice cold 3 M sodium acetate (pH 5.5). Carry out these washes by thoroughly resuspending the pellet with a disposable plastic rod in 3 M sodium acetate, incubating on ice for 10-30 min, and centrifuging at 10,000 x g for 15-20 min. Dissolve the final pellet completely in water, and precipitate high molecular weight RNA from a mixture of 0.2 M sodium acetate and 2.5 volumes of ethanol. Remove any remaining DNA by digesting with RNase-free DNase in 10 mM Tris-HCl (pH 7.5)-10 mM magnesium chloride-100 mM sodium chloride, 5 mM vanadyl ribonucleoside complex-50 units/ml RNase-free DNase for 30 min at 37°C (this cleanup is not necessary when dealing with small amounts of tissue). Extract the RNA twice with PCIA (see A9) and then precipitate as described. Dissolve the final pellet in water and quantitate at an absorbance of A260. The yield of RNA is approximately 1 mg/g of tissue with an A260/A280 ratio of at least 1.8. When virus is added to tissue before RNA isolation, virus particles are added to 0.5 ml of a 10% homogenate of tissue in extraction buffer only. RNA is then isolated as described above by phenol extraction, except that all extractions are done in Eppendorf tubes. After adding an equal volume of phenol, vortex the tubes for 1 min and centrifuge to separate the phases. Repeat the phenol extraction once. Carry out the 3 M sodium acetate extractions with 0.5 ml of solution and omit the DNAse digestion (2).
-
Reverse transcription (RT)
Carry out RT of RNA in 20 µl reaction mixtures containing 50 mM Tris HCl (pH 8.3), 75 mM KCl, 10 mM MgCl2, 1 mM concentrations of each of the four deoxyribonucleoside triphosphates (dNTPs), 10 mM dithiothreitol, 1 unit of human placental RNAse inhibitor per µl, 0.5 µg oligo(dT)16, 15 units of avian myeloblastosis virus reverse transcriptase, and indicated amounts of seeded or unseeded tissue RNA and competitor template RNA. Incubate reactions at 22°C for 10 min followed by 50 min at 42°C, 5 min at 99°C, and 5 min at 4°C. Centrifuge the reaction tubes for 5 min at full speed in an Eppendorf centrifuge (2).
-
PCR
Perform PCR amplification in 50 µl volumes, which contain 5-10 µl cDNA pool from the RT reactions, 3 mM mgCl2, 200 µM of each dNTPs, 0.5 µM of each amplification primer, and 1.5 units of Taq DNA polymerase. Denature the mixture initially for 3 min at 95°C, followed by 35-40 cycles of amplification, each consisting of 90 s at 94°C, 90 s at 63°C, and 120 s at 72°C. Perform a final extension at 72°C for 10 min; analyze 20 µl of PCR reaction by gel electrophoresis. When quantitation is desired, perform the PCR reaction in the presence of 5 µCi per reaction of [-32P]dCTP in addition to unlabeled dCTP. Separate the products by gel electrophoresis, and excise and count bands corresponding to the wild-type and competitor template in a liquid scintillation counter (1).
-
Quantitation of viral RNA molecules
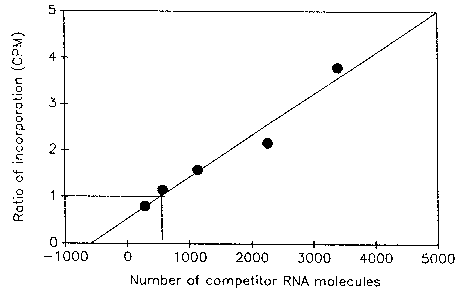
Figure 1 shows the results of an experiment to quantitate the number of viral RNA molecules in a crude virus preparation obtained from the Centers for Disease Control and Prevention, Atlanta, GA. To avoid losses during RNA isolation, crude virus preparation was diluted 500-fold in RNase-free water and heated at 95°C for 5 min to dissociate RNA-protein complexes and then chilled on ice. A 1 µl aliquot of heated virus was then mixed with several concentrations of competitor RNA and reverse transcribed. From each cDNA pool, 5 µl was then amplified by PCR as described above, except that 32P dATP was used as the label. Products were separated in a 3% NuSieve-1% agarose gel. Bands corresponding to wild-type and competitor PCR products were cut out and counted. Incorporation into PCR product generated from competitor RNA was corrected for the loss of A and T residues caused by deletion of 63 base pairs. The ratio of incorporation in the two products was then plotted against the number of competitor RNA molecules added and a linear regression graph was obtained. The number of viral RNA molecules in the crude virus preparation is the same as the number of competitor RNA molecules added when the ratio of incorporation in the two PCR products is equal to one. Based on the results of the experiment, the number of viral RNA molecules in the virus preparation was estimated to be 2.5 x 108/ml.
Figure 1. Quantitation of viral RNA molecules in a crude virus preparation by competitive PCR. A fixed concentration of heated virus was mixed with increasingly higher numbers of a competitor RNA molecule and reverse transcribed. A portion of the cDNA pool was then amplified as described in the text.
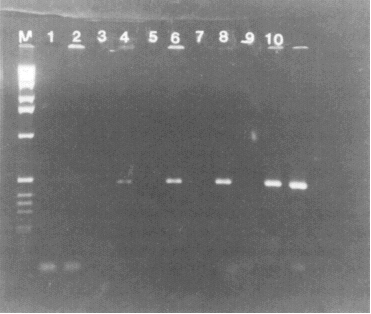
Figure 2. Detection of HAV sequence in RNA isolated from clam tissue seeded with wild-type HAV. RNA from seeded and unseeded tissue was isolated as described in the text. RNA representing 0-2500 particles of virus was then reverse transcribed and one-fifth of each cDNA pool was then amplified. Lanes 1 and 2, RNA isolated from unseeded tissue; lanes 3 and 4, RNA representing 50 particles of virus in the RT mixture; lanes 5 and 6, 250 particles of virus; lanes 7 and 8, 500 particles of virus; lanes 9 and 10, 2500 particles of virus; lane M, 1-kb DNA marker. Reverse transcriptase was omitted from the reaction mixtures in lanes 1, 3, 5, 7, and 9.
To detect HAV RNA in Mercenaria mercenaria seeded with wild-type HAV, RNA was isolated from 50 mg tissue seeded with 500 or 5000 particles of virus (based on the number of viral RNA molecules estimated from Fig. 1). As control, RNA was also isolated from tissue that had not been seeded with HAV. All RNA samples were then reverse transcribed and amplified as described above except that radioactive deoxynucleotide and competitor RNA were omitted. PCR products were analyzed by 1.6% agarose gel. As shown in Fig. 2, viral sequences were readily detected in samples seeded with virus before RNA isolation but were not detectable in the unseeded sample. No viral sequences were detected when reverse transcriptase was omitted from the reaction mixture. In subsequent studies, it was estimated that this method can detect 2000 virus particles per gram of shellfish tissue. The minimum infectious dose is unknown, but presumably is less than this number. The lack of requirement of any special reagents such as specific antibodies should make this method generally applicable for the detection of HAV.
References
- Goswami, B.B., W.H. Koch, and T.A. Cebula, T.A. 1994. Competitor template RNA for detection and quantitation of hepatitis A virus by PCR. BioTechniques 16:114-121.
- Goswami, B.B., W.H. Koch, and T.A. Cebula. 1993. Detection of hepatitis A virus in Mercenaria mercenaria by coupled reverse transcription and polymerase chain reaction. Appl. Environ. Microbiol. 59:2765-2770.
Hypertext Source: Bacteriological Analytical Manual, 8th Edition, Revision A, 1998. Chapter 26 .