
Master Files
Introduction to Master Files for Devices (MAFs)
A premarket approval application (PMA) or an investigational device exemption application (IDE) usually contains data and other information that the applicant has developed and regards as trade secret or confidential commercial financial information. Often the applicant needs to use another party's product (e.g., ingredient, subassembly, or accessory) or facility in the manufacture of the device. In order that a sound scientific evaluation may be made of the PMA, IDE, or other device submission, the review of data and other information related to the other party's product, facility, or manufacturing procedures is required. The other party, while willing to allow FDA's confidential review of this information, may not want the IDE, premarket notification [510(k)], or PMA applicant to have direct access to the information. To help preserve the trade secrets of the ancillary medical device industry and at the same time facilitate the sound scientific evaluation of medical devices, FDA established the device master file system. In addition, a master file may be considered when several applications may be submitted for different products which may use a common material or process, etc., such as the same sterilization method.
This guideline only applies to the master files (MAFs) submitted to the Center for Devices and Radiological Health (CDRH). Master files in support of other products regulated by FDA, even though they may contain information previously submitted in an MAF, are to be submitted to the appropriate FDA center(s). The content and the way the master file is used may vary among FDA centers.
Other master files submitted for review in support of documents filed with FDA are:
- Biologics Master Files supporting Notices of Claimed Investigational Exemption for a New Drug (INDs) for biologics and biologic licenses. For more information please refer to CBER’s Drug Master Files for CBER-Regulated Products webpage.
- Drug Master Files (DMFs) supporting Investigational New Drug Applications (INDs), New Drug Applications (NDAs), and Abbreviated New Drug Applications (ANDAs). For more information please refer to CDER’s Drug Master Files webpage.
- Food Master Files (FMFs) supporting Food Additive and Color Additive Petitions. For more please refer to CFSAN’s Food Master Files webpage.
- Veterinary Medicine Master Files supporting Investigational New Animal Exemptions (INADs) and New Animal Drug Applications (NADAs). For more information please refer to the CVM’s Veterinary Medicine Master Files webpage.
The following are commonly used terms relating to MAFs for devices and the documents referring to them:
- An "MAF holder" is an organization or person filing an MAF.
- An "applicant" is an organization or person filing a PMA.
- A "sponsor" is an organization or person filing an IDE.
- An "agent or representative for an MAF holder" is a person or organization authorized to represent the MAF holder before FDA concerning the contents of the MAF.
Types of MAFs and Their Functions
Usually, MAFs are accepted from those organizations or persons who have not submitted or will not directly submit the information in a PMA, IDE, 510(k), or other device-related submission to FDA for the reasons identified above. MAFs may be submitted for various functions. These functions have been grouped by the following types:
- facilities and manufacturing procedures and controls;
- synthesis, formulation, purification and specifications for chemicals, materials (e.g., an alloy, plastic, etc.) or subassemblies for a device;
- packaging materials;
- contract packaging and other manufacturing (e.g., sterilization);
- nonclinical study data; and
- clinical study data.
- In the first volume of each copy, include a signed and dated cover letter identifying the submission as a device master file (MAF) and briefly describe the subject of the submission.
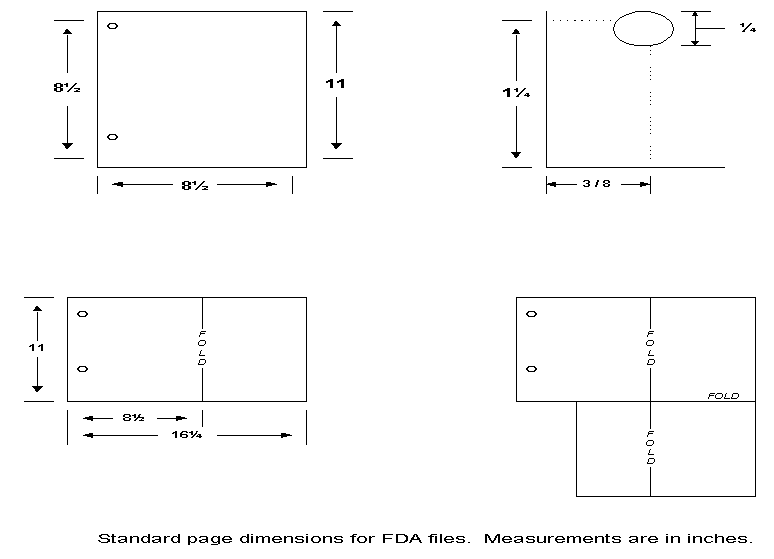
- U.S. standard size and weight bond paper (8 inches x 11 inches) is preferred. Page sizes should not exceed 11 inches in length and 8 inches in width.
- FDA's system for storing MAFs requires that the volume be bound on the left side of the page using the U.S. standard size loose leaf page.
- Should it be necessary to use individual pages larger than the U.S. standard page size to present a floor diagram, device design, electrical diagram, etc., those pages should be mounted to allow the page to be opened for review and folded without damage when the volume is shelved.
- Allow a left margin of at least 3/4 inch to assure that the text is clear of binding.
- Individual volumes should not exceed 2 inches in thickness.
- DO NOT BIND volumes in book form or with plastic or metal spirals.
The Content of an MAF
There are no specific content requirements for a MAF. However, a submission will not be accepted as an MAF if it is not substantive in nature and does not contain information that may reasonably be regarded as trade secret or confidential commercial or financial information.
The submission must include a cover letter, preferably bearing company letterhead, signed by a responsible official (e.g., Director of Regulatory Affairs or another manager). The letter should identify the submission as an MAF, and a contact person at the company or designated agent should be listed.
An MAF must be in the English language or be accompanied by accurate English translations of any of the documents that are in a language other than English.
Amending an MAF
After submission of an MAF, its information may need to be updated as a result of additional testing, additional applications of the MAF information or modification of the product that is the subject of the MAF.
Changes made in product or manufacturing operations can affect a client's medical device and possibly result in the client marketing a device differing from that originally approved by FDA. This can also have an effect on product liability obligations and a client's compliance with applicable FDA laws and regulations. It is, therefore, necessary to notify a client before proposed changes are made in your operations or product.
Authorization to an MAF
Information in an MAF may be incorporated by reference in a client's PMA, 510(k), or IDE, or other submissions to FDA. Their use of information in an MAF can only be authorized by the MAF holder or by a designated agent if so authorized. This authorization must be on company letterhead or that of the agent or representative. After FDA has referred to an MAF and the client's application has been approved, authorization cannot be withdrawn.
An MAF holder should provide a letter of authorization directly to a client with instructions that: (1) the original of the authorization letter be included in the original copy of the client's submission and (2) a copy be placed in each subsequent copy of the client's submission. An authorization letter should not be sent directly to CDRH for inclusion in the MAF or the client's submission.
Below is the format for a sample authorization letter.
[Use company letterhead stationery giving company name, address, and telephone number.1
[Date]
[Client's name]
[Mailing address including City, State and Zip Code]
Dear [Person responsible for filing the client's application]:
This letter authorizes the Food and Drug Administration to include by reference information in our device master file, MAF #[specify the correct MAF number] for [specify the subject of the MAF, e.g., PMA, IDE or 510(k)] in [specify the client's device by generic or trade name]. [You may add further restricting wording if you desire].
Sincerely yours,
[Name and title of the person responsible for MAF authorizations]
Good Manufacturing Practice Regulations for Medical Devices
Organizations or persons who submit IDEs, 510(k)s, or PMAs, or other device-related submissions to FDA may use contract manufacturers, sterilizers, packagers, etc., in the manufacture of their devices. The latter, for trade secret or confidentiality purposes, may submit a description of its facilities, manufacturing procedures and processes, and quality control procedures in an MAF rather than provide this information directly to the client for inclusion in its submission.
In their evaluation of the client's submission, FDA may inspect the MAF holder's facilities and manufacturing operations. When the MAF holder's operation is subject to the Good Manufacturing Practice (GMP) regulations for medical devices (21 CFR 820), the MAF must address all appropriate GMP requirements applicable to the MAF holder's operation. A client's submission may be adversely affected if the MAF is incomplete or inaccurate. This is especially true in the case of a PMA because of the statutory requirement that a PMA contain a full description of the methods used in, and the facilities and controls used for, the manufacture, processing, and when relevant, packing and installation of the device.
Representing an MAF
If an MAF submitter is a foreign company, FDA recommends that it retain an agent or representative in the United States. This will usually facilitate any clarification or correction of deficiencies in the MAF information. Identify any agent by name, address, and telephone number and specify any limitations in the authority of the agent or representative. If limitations are not specified, FDA will assume that all information in the MAF may be discussed with the agent or representative. A designated agent or representative can be added or removed only by amending the MAF.
Where to Submit an MAF
An MAF and all amendments should be submitted to the appropriate Center’s Document Control Center (DCC). The current mailing address for CDRH’s DCC and a link to CBER’s DCC’s mailing address are provided on the eCopy Program for Medical Device Submissions webpage.
SPECIAL NOTE: FDA will not pay any shipping or C.O.D. charges for incoming submissions or pick-up MAFs from the carrier. It is the submitter's responsibility to assure prepaid delivery to the above address.
Receipt will be acknowledged by letter of all new MAF submissions and amendments. FDA's acknowledgment letter of the original MAF will include the FDA assigned MAF reference number, which must be included in all amendments to the MAF.
Freedom of Information Act and an MAF
Under the Freedom of Information Act (FOI), information in 510(k)s, IDEs, PMAs and other device-related submissions, such as MAFs, is subject to public disclosure unless determined by FDA to be trade secret or confidential commercial or financial information within the meaning of 21 CFR 20.61 or otherwise prohibited from public disclosure. In the case of PMAs, FDA is required to make publicly available a detailed summary of the safety and effectiveness data which is the basis for the FDA decision to approve, or deny approval of, the PMA. Information in such summaries cannot be used to establish the safety or effectiveness for another device by any person other than the one who submitted the information.
FOI public disclosure provisions apply whether or not the information in the 510(k), IDE, PMA, or other device-related submission was submitted by the applicant or is included by authorized reference to an MAF.
MAF holders should identify information in their MAFs which they consider to be trade secret or confidential commercial/financial information within the meaning of 21 CFR 20.61. MAF information already in the public domain is subject to disclosure (e.g., any published literature, catalog, or product specification sheets distributed by the MAF holder to potential customers).
CDRH Contact Office for Assistance
Procedural and other questions regarding MAFs should be directed to:
- Wanda.sawyer-major@fda.hhs.gov or 301-796-6568