biochemistry
Finding HIV’s ‘Sweet Spot’
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Each year, about 30,000 people in the United States contract the human immunodeficiency virus (HIV), the cause of AIDS [1]. Thankfully, most can control their HIV infections with antiretroviral therapy and will lead productive, high-quality lives. Many will even reach a point where they have no detectable levels of virus circulating in their blood. However, all must still worry that the undetectable latent virus hidden in their systems could one day reactivate and lead to a range of serious health complications.
Now, an NIH-funded team has found that patterns of sugars at the surface of our own human immune cells affect their vulnerability to HIV infection. These data suggest it may be possible to find the infected immune cells harboring the last vestiges of virus by reading the sugar profiles on their surfaces. If so, it would move us a step closer to eliminating latent HIV infection and ultimately finding a cure for this horrible virus.
These fascinating new findings come from a team led by Nadia Roan, Gladstone Institutes, San Francisco and Mohamed Abdel-Mohsen, The Wistar Institute, Philadelphia, PA. Among its many areas of study, the Roan lab is interested in why HIV favors infecting specific subsets of a special type of immune cell called memory CD4 T cells. These cells come in different varieties. They also play important roles in the immune system’s ability to recall past infections and launch a rapid response to an emerging repeat infection.
For years, her team and others have tried to understand the interplay between HIV and human immune cells primarily by studying the proteins present at the cell surface. But living cells and their proteins also are coated in sugars and, the presence or absence of these carbohydrates is essential to their biochemistry.
In the new study, published in the journal eLife, the researchers included for the first time the patterns of these sugars in their study of cell surface proteins [2]. They, like many labs, hadn’t done so previously for technical reasons: it’s much easier to track these proteins than sugars.
To overcome this technical hurdle, Roan’s team turned to an approach that it uses for quantifying levels of proteins on the surface of single cells. The method, called CyTOF, uses metal-studded antibodies that stick to proteins, uniquely marking precise patterns of selected proteins, in this case, on individual HIV-infected cells.
In collaboration with Abdel-Mohsen, a glycobiology expert, they adapted this method for cell surface sugars. They did it by adding molecules called lectins, which stick to sugar molecules with specific shapes and compositions.
With this innovation, Roan and team report that they learned to characterize and quantify levels of 34 different proteins on the cell surface simultaneously with five types of sugars. Their next questions were: Could those patterns of cell-surface sugars help them differentiate between different types of immune cells? If so, might those patterns help to define a cell’s susceptibility to HIV?
The answer appears to be yes to both questions. Their studies revealed tremendous diversity in the patterns of sugars at the cells surfaces. Those patterns varied depending on a cell’s tissue of origin—in this case, from blood, tonsil, or the reproductive tract. The patterns also varied depending on the immune cell type—memory CD4 T cells versus other T cells or antibody-producing B cells.
Those sugar and protein profiles offered important clues as to which cells HIV prefers to infect. More specifically, compared to uninfected memory CD4 T cells, the infected ones had higher surface levels of two sugars, known as fucose [3] and sialic acid [4]. What’s more, during HIV infection, levels of both sugars increased.
Scientists already knew that HIV changes the proteins that the infected memory CD4 T cell puts on its surface, a process known as viral remodeling. Now it appears that something similar happens with sugars, too. The new findings suggest the virus increases levels of sialic acid at the cell surface in ways that may help the virus to survive. That’s especially intriguing because sialic acid also is associated with a cell’s ability to avoid detection by the immune system.
The Roan and Abdel-Mohsen labs now plan to team up again to apply their new method to study latent infection. They want to find sugar-based patterns that define those lingering infected cells and see if it’s possible to target them and eliminate the lingering HIV.
What’s also cool is this study indicates that by performing single-cell analyses and sorting cells based on their sugar and protein profiles, it may be possible to discover distinct new classes of immune and other cells that have eluded earlier studies. As was the case with HIV, this broader protein-sugar profile could hold the key to gaining deeper insights into disease processes throughout the body.
References:
[1] Diagnoses of HIV infection in the United States and dependent areas, 2020. HIV Surveillance Report, May 2020; 33; Centers for Disease Control and Prevention.
[2] Single-cell glycomics analysis by CyTOF-Lec reveals glycan features defining cells differentially susceptible to HIV. Ma T, McGregor M, Giron L, Xie G, George AF, Abdel-Mohsen M, Roan NR.eLife 2022 July 5;11:e78870
[3] Biological functions of fucose in mammals. Schneider M, Al-Shareffi E, Haltiwanger RS. Glycobiology. 2016 Jun;26(6):543.
[4] Sialic acids and other nonulosonic acids. Lewis AL, Chen X, Schnaar RL, Varki A. In Essentials of Glycobiology [Internet]. 4th edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2022.
Links:
HIV/AIDS (National Institute of Allergy and Infectious Diseases/NIH)
Roan Lab (University of California, San Francisco)
Mohamed Abdel-Mohsen (The Wistar Institute, Philadelphia, PA)
NIH Support: National Institute of Allergy and Infectious Diseases; National Institute of Diabetes and Digestive and Kidney Diseases; National Institute on Aging; National Institute of Neurological Disorders and Stroke
How One Change to The Coronavirus Spike Influences Infectivity
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Since joining NIH, I’ve held a number of different leadership positions. But there is one position that thankfully has remained constant for me: lab chief. I run my own research laboratory at NIH’s National Institute of Dental and Craniofacial Research (NIDCR).
My lab studies a biochemical process called O-glycosylation. It’s fundamental to life and fascinating to study. Our cells are often adorned with a variety of carbohydrate sugars. O-glycosylation refers to the biochemical process through which these sugar molecules, either found at the cell surface or secreted, get added to proteins. The presence or absence of these sugars on certain proteins plays fundamental roles in normal tissue development and first-line human immunity. It also is associated with various diseases, including cancer.
Our lab recently joined a team of NIH scientists led by my NIDCR colleague Kelly Ten Hagen to demonstrate how O-glycosylation can influence SARS-CoV-2, the coronavirus that causes COVID-19, and its ability to fuse to cells, which is a key step in infecting them. In fact, our data, published in the journal Proceedings of the National Academy of Sciences, indicate that some variants, seem to have mutated to exploit the process to their advantage [1].
The work builds on the virus’s reliance on the spike proteins that crown its outer surface to attach to human cells. Once there, the spike protein must be activated to fuse and launch an infection. That happens when enzymes produced by our own cells make a series of cuts, or cleavages, to the spike protein.
The first cut comes from an enzyme called furin. We and others had earlier evidence that O-glycosylation can affect the way furin makes those cuts. That got us thinking: Could O-glycosylation influence the interaction between furin and the spike protein? The furin cleavage area of the viral spike was indeed adorned with sugars, and their presence or absence might influence spike activation by furin.
We also noticed the Alpha and Delta variants carry a mutation that removes the amino acid proline in a specific spot. That was intriguing because we knew from earlier work that enzymes called GALNTs, which are responsible for adding bulky sugar molecules to proteins, prefer prolines near O-glycosylation sites.
It also suggested that loss of proline in the new variants could mean decreased O-glycosylation, which might then influence the degree of furin cleavage and SARS-CoV-2’s ability to enter cells. I should note that the recent Omicron variant was not examined in the current study.
After detailed studies in fruit fly and mammalian cells, we demonstrated in the original SARS-CoV-2 virus that O-glycosylation of the spike protein decreases furin cleavage. Further experiments then showed that the GALNT1 enzyme adds sugars to the spike protein and this addition limits the ability of furin to make the needed cuts and activate the spike protein.
Importantly, the spike protein change found in the Alpha and Delta variants lowers GALNT1 activity, making it easier for furin to start its activating cuts. It suggests that glycosylation of the viral spike by GALNT1 may limit infection with the original virus, and that the Alpha and Delta variant mutation at least partially overcomes this effect, to potentially make the virus more infectious.
Building on these studies, our teams looked for evidence of GALNT1 in the respiratory tracts of healthy human volunteers. We found that the enzyme is indeed abundantly expressed in those cells. Interestingly, those same cells also express the ACE2 receptor, which SARS-CoV-2 depends on to infect human cells.
It’s also worth noting here that the Omicron variant carries the very same spike mutation that we studied in Alpha and Delta. Omicron also has another nearby change that might further alter O-glycosylation and cleavage of the spike protein by furin. The Ten Hagen lab is looking into these leads to learn how this region in Omicron affects spike glycosylation and, ultimately, the ability of this devastating virus to infect human cells and spread.
Reference:
[1] Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Zhang L, Mann M, Syed Z, Reynolds HM, Tian E, Samara NL, Zeldin DC, Tabak LA, Ten Hagen KG. PNAS. 2021 Nov 23;118(47).
Links:
COVID-19 Research (NIH)
Kelly Ten Hagen (National Institute of Dental and Craniofacial Research/NIH)
Lawrence Tabak (NIDCR)
NIH Support: National Institute of Dental and Craniofacial Research
Biomedical Research Leads Science’s 2021 Breakthroughs
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Hi everyone, I’m Larry Tabak. I’ve served as NIH’s Principal Deputy Director for over 11 years, and I will be the acting NIH director until a new permanent director is named. In my new role, my day-to-day responsibilities will certainly increase, but I promise to carve out time to blog about some of the latest research progress on COVID-19 and any other areas of science that catch my eye.
I’ve also invited the directors of NIH’s Institutes and Centers (ICs) to join me in the blogosphere and write about some of the cool science in their research portfolios. I will publish a couple of posts to start, then turn the blog over to our first IC director. From there, I envision alternating between posts from me and from various IC directors. That way, we’ll cover a broad array of NIH science and the tremendous opportunities now being pursued in biomedical research.
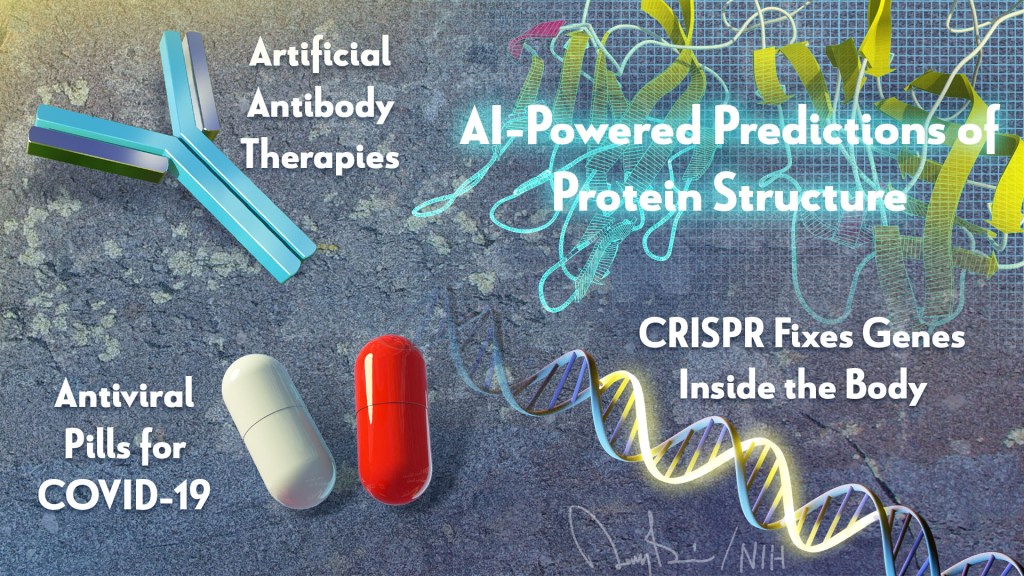
Since I’m up first, let’s start where the NIH Director’s Blog usually begins each year: by taking a look back at Science’s Breakthroughs of 2021. The breakthroughs were formally announced in December near the height of the holiday bustle. In case you missed the announcement, the biomedical sciences accounted for six of the journal Science’s 10 breakthroughs. Here, I’ll focus on four biomedical breakthroughs, the ones that NIH has played some role in advancing, starting with Science’s editorial and People’s Choice top-prize winner:
Breakthrough of the Year: AI-Powered Predictions of Protein Structure
The biochemist Christian Anfinsen, who had a distinguished career at NIH, shared the 1972 Nobel Prize in Chemistry, for work suggesting that the biochemical interactions among the amino acid building blocks of proteins were responsible for pulling them into the final shapes that are essential to their functions. In his Nobel acceptance speech, Anfinsen also made a bold prediction: one day it would be possible to determine the three-dimensional structure of any protein based on its amino acid sequence alone. Now, with advances in applying artificial intelligence to solve biological problems—Anfinsen’s bold prediction has been realized.
But getting there wasn’t easy. Every two years since 1994, research teams from around the world have gathered to compete against each other in developing computational methods for predicting protein structures from sequences alone. A score of 90 or above means that a predicted structure is extremely close to what’s known from more time-consuming work in the lab. In the early days, teams more often finished under 60.
In 2020, a London-based company called DeepMind made a leap with their entry called AlphaFold. Their deep learning approach—which took advantage of 170,000 proteins with known structures—most often scored above 90, meaning it could solve most protein structures about as well as more time-consuming and costly experimental protein-mapping techniques. (AlphaFold was one of Science’s runner-up breakthroughs last year.)
This year, the NIH-funded lab of David Baker and Minkyung Baek, University of Washington, Seattle, Institute for Protein Design, published that their artificial intelligence approach, dubbed RoseTTAFold, could accurately predict 3D protein structures from amino acid sequences with only a fraction of the computational processing power and time that AlphaFold required [1]. They immediately applied it to solve hundreds of new protein structures, including many poorly known human proteins with important implications for human health.
The DeepMind and RoseTTAFold scientists continue to solve more and more proteins [1,2], both alone and in complex with other proteins. The code is now freely available for use by researchers anywhere in the world. In one timely example, AlphaFold helped to predict the structural changes in spike proteins of SARS-CoV-2 variants Delta and Omicron [3]. This ability to predict protein structures, first envisioned all those years ago, now promises to speed fundamental new discoveries and the development of new ways to treat and prevent any number of diseases, making it this year’s Breakthrough of the Year.
Anti-Viral Pills for COVID-19
The development of the first vaccines to protect against COVID-19 topped Science’s 2020 breakthroughs. This year, we’ve also seen important progress in treating COVID-19, including the development of anti-viral pills.
First, there was the announcement in October of interim data from Merck, Kenilworth, NJ, and Ridgeback Biotherapeutics, Miami, FL, of a significant reduction in hospitalizations for those taking the anti-viral drug molnupiravir [4] (originally developed with an NIH grant to Emory University, Atlanta). Soon after came reports of a Pfizer anti-viral pill that might target SARS-CoV-2, the novel coronavirus that causes COVID-19, even more effectively. Trial results show that, when taken within three days of developing COVID-19 symptoms, the pill reduced the risk of hospitalization or death in adults at high risk of progressing to severe illness by 89 percent [5].
On December 22, the Food and Drug Administration (FDA) granted Emergency Use Authorization (EUA) for Pfizer’s Paxlovid to treat mild-to-moderate COVID-19 in people age 12 and up at high risk for progressing to severe illness, making it the first available pill to treat COVID-19 [6]. The following day, the FDA granted an EUA for Merck’s molnupiravir to treat mild-to-moderate COVID-19 in unvaccinated, high-risk adults for whom other treatment options aren’t accessible or recommended, based on a final analysis showing a 30 percent reduction in hospitalization or death [7].
Additional promising anti-viral pills for COVID-19 are currently in development. For example, a recent NIH-funded preclinical study suggests that a drug related to molnupiravir, known as 4’-fluorouridine, might serve as a broad spectrum anti-viral with potential to treat infections with SARS-CoV-2 as well as respiratory syncytial virus (RSV) [8].
Artificial Antibody Therapies
Before anti-viral pills came on the scene, there’d been progress in treating COVID-19, including the development of monoclonal antibody infusions. Three monoclonal antibodies now have received an EUA for treating mild-to-moderate COVID-19, though not all are effective against the Omicron variant [9]. This is also an area in which NIH’s Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) public-private partnership has made big contributions.
Monoclonal antibodies are artificially produced versions of the most powerful antibodies found in animal or human immune systems, made in large quantities for therapeutic use in the lab. Until recently, this approach had primarily been put to work in the fight against conditions including cancer, asthma, and autoimmune diseases. That changed in 2021 with success using monoclonal antibodies against infections with SARS-CoV-2 as well as respiratory syncytial virus (RSV), human immunodeficiency virus (HIV), and other infectious diseases. This earned them a prominent spot among Science’s breakthroughs of 2021.
Monoclonal antibodies delivered via intravenous infusions continue to play an important role in saving lives during the pandemic. But, there’s still room for improvement, including new formulations highlighted on the blog last year that might be much easier to deliver.
CRISPR Fixes Genes Inside the Body
One of the most promising areas of research in recent years has been gene editing, including CRISPR/Cas9, for fixing misspellings in genes to treat or even cure many conditions. This year has certainly been no exception.
CRISPR is a highly precise gene-editing system that uses guide RNA molecules to direct a scissor-like Cas9 enzyme to just the right spot in the genome to cut out or correct disease-causing misspellings. Science highlights a small study reported in The New England Journal of Medicine by researchers at Intellia Therapeutics, Cambridge, MA, and Regeneron Pharmaceuticals, Tarrytown, NY, in which six people with hereditary transthyretin (TTR) amyloidosis, a condition in which TTR proteins build up and damage the heart and nerves, received an infusion of guide RNA and CRISPR RNA encased in tiny balls of fat [10]. The goal was for the liver to take them up, allowing Cas9 to cut and disable the TTR gene. Four weeks later, blood levels of TTR had dropped by at least half.
In another study not yet published, researchers at Editas Medicine, Cambridge, MA, injected a benign virus carrying a CRISPR gene-editing system into the eyes of six people with an inherited vision disorder called Leber congenital amaurosis 10. The goal was to remove extra DNA responsible for disrupting a critical gene expressed in the eye. A few months later, two of the six patients could sense more light, enabling one of them to navigate a dimly lit obstacle course [11]. This work builds on earlier gene transfer studies begun more than a decade ago at NIH’s National Eye Institute.
Last year, in a research collaboration that included former NIH Director Francis Collins’s lab at the National Human Genome Research Institute (NHGRI), we also saw encouraging early evidence in mice that another type of gene editing, called DNA base editing, might one day correct Hutchinson-Gilford Progeria Syndrome, a rare genetic condition that causes rapid premature aging. Preclinical work has even suggested that gene-editing tools might help deliver long-lasting pain relief. The technology keeps getting better, too. This isn’t the first time that gene-editing advances have landed on Science’s annual Breakthrough of the Year list, and it surely won’t be the last.
The year 2021 was a difficult one as the pandemic continued in the U.S. and across the globe, taking far too many lives far too soon. But through it all, science has been relentless in seeking and finding life-saving answers, from the rapid development of highly effective COVID-19 vaccines to the breakthroughs highlighted above.
As this list also attests, the search for answers has progressed impressively in other research areas during these difficult times. These groundbreaking discoveries are something in which we can all take pride—even as they encourage us to look forward to even bigger breakthroughs in 2022. Happy New Year!
References:
[1] Accurate prediction of protein structures and interactions using a three-track neural network. Baek M, DiMaio F, Anishchenko I, Dauparas J, Grishin NV, Adams PD, Read RJ, Baker D., et al. Science. 2021 Jul 15:eabj8754.
[2] Highly accurate protein structure prediction with AlphaFold. Jumper J, Evans R, Pritzel A, Green T, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. et al. Nature. 2021 Jul 15.
[3] Structural insights of SARS-CoV-2 spike protein from Delta and Omicron variants. Sadek A, Zaha D, Ahmed MS. preprint bioRxiv. 2021 Dec 9.
[5] Pfizer’s novel COVID-19 oral antiviral treatment candidate reduced risk of hospitalization or death by 89% in interim analysis of phase 2/3 EPIC-HR Study. Pfizer. 5 November 52021.
[6] Coronavirus (COVID-19) Update: FDA authorizes first oral antiviral for treatment of COVID-19. Food and Drug Administration. 22 Dec 2021.
[7] Coronavirus (COVID-19) Update: FDA authorizes additional oral antiviral for treatment of COVID-19 in certain adults. Food and Drug Administration. 23 Dec 2021.
[8] 4′-Fluorouridine is an oral antiviral that blocks respiratory syncytial virus and SARS-CoV-2 replication. Sourimant J, Lieber CM, Aggarwal M, Cox RM, Wolf JD, Yoon JJ, Toots M, Ye C, Sticher Z, Kolykhalov AA, Martinez-Sobrido L, Bluemling GR, Natchus MG, Painter GR, Plemper RK. Science. 2021 Dec 2.
[9] Anti-SARS-CoV-2 monoclonal antibodies. NIH COVID-19 Treatment Guidelines. 16 Dec 2021.
[10] CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O’Connell D, Walsh KR, Wood K, Phillips J, Xu Y, Amaral A, Boyd AP, Cehelsky JE, McKee MD, Schiermeier A, Harari O, Murphy A, Kyratsous CA, Zambrowicz B, Soltys R, Gutstein DE, Leonard J, Sepp-Lorenzino L, Lebwohl D. N Engl J Med. 2021 Aug 5;385(6):493-502.
[11] Editas Medicine announces positive initial clinical data from ongoing phase 1/2 BRILLIANCE clinical trial of EDIT-101 For LCA10. Editas Medicine. 29 Sept 2021.
Links:
Structural Biology (National Institute of General Medical Sciences/NIH)
The Structures of Life (NIGMS)
COVID-19 Research (NIH)
2021 Science Breakthrough of the Year (American Association for the Advancement of Science, Washington, D.C)
Enlisting Monoclonal Antibodies in the Fight Against COVID-19
Posted on by Dr. Francis Collins

We now know that the immune system of nearly everyone who recovers from COVID-19 produces antibodies against SARS-CoV-2, the novel coronavirus that causes this easily transmitted respiratory disease [1]. The presence of such antibodies has spurred hope that people exposed to SARS-CoV-2 may be protected, at least for a time, from getting COVID-19 again. But, in this post, I want to examine another potential use of antibodies: their promise for being developed as therapeutics for people who are sick with COVID-19.
In a recent paper in the journal Science, researchers used blood drawn from a COVID-19 survivor to identify a pair of previously unknown antibodies that specifically block SARS-CoV-2 from attaching to human cells [2]. Because each antibody locks onto a slightly different place on SARS-CoV-2, the vision is to use these antibodies in combination to block the virus from entering cells, thereby curbing COVID-19’s destructive spread throughout the lungs and other parts of the body.
The research team, led by Yan Wu, Capital Medical University, Beijing, first isolated the pair of antibodies in the laboratory, starting with white blood cells from the patient. They were then able to produce many identical copies of each antibody, referred to as monoclonal antibodies. Next, these monoclonal antibodies were simultaneously infused into a mouse model that had been infected with SARS-CoV-2. Just one infusion of this combination antibody therapy lowered the amount of viral genetic material in the animals’ lungs by as much as 30 percent compared to the amount in untreated animals.
Monoclonal antibodies are currently used to treat a variety of conditions, including asthma, cancer, Crohn’s disease, and rheumatoid arthritis. One advantage of this class of therapeutics is that the timelines for their development, testing, and approval are typically shorter than those for drugs made of chemical compounds, called small molecules. Because of these and other factors, many experts think antibody-based therapies may offer one of the best near-term options for developing safe, effective treatments for COVID-19.
So, what exactly led up to this latest scientific achievement? The researchers started out with a snippet of SARS-CoV-2’s receptor binding domain (RBD), a vital part of the spike protein that protrudes from the virus’s surface and serves to dock the virus onto an ACE2 receptor on a human cell. In laboratory experiments, the researchers used the RBD snippet as “bait” to attract antibody-producing B cells in a blood sample obtained from the COVID-19 survivor. Altogether, the researchers identified four unique antibodies, but two, which they called B38 and H4, displayed a synergistic action in binding to the RBD that made them stand out for purposes of therapeutic development and further testing.
To complement their lab and animal experiments, the researchers used a particle accelerator called a synchrotron to map, at near-atomic resolution, the way in which the B38 antibody locks onto its viral target. This structural information helps to clarify the precise biochemistry of the complex interaction between SARS-CoV-2 and the antibody, providing a much-needed guide for the rational design of targeted drugs and vaccines. While more research is needed before this or other monoclonal antibody therapies can be used in humans suffering from COVID-19, the new work represents yet another example of how basic science is expanding fundamental knowledge to advance therapeutic discovery for a wide range of health concerns.
Meanwhile, there’s been other impressive recent progress towards the development of monoclonal antibody therapies for COVID-19. In work described in the journal Nature, an international research team started with a set of neutralizing antibodies previously identified in a blood sample from a person who’d recovered from a different coronavirus-caused disease, called severe acute respiratory syndrome (SARS), in 2003 [3]. Through laboratory and structural imaging studies, the researchers found that one of these antibodies, called S309, proved particularly effective at neutralizing the coronavirus that causes COVID-19, SARS-CoV-2, because of its potent ability to target the spike protein that enables the virus to enter cells. The team, which includes NIH grantees David Veesler, University of Washington, Seattle, and Davide Corti, Humabs Biomed, a subsidiary of Vir Biotechnology, has indicated that S309 is already on an accelerated development path toward clinical trials.
In the U.S. and Europe, the Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) partnership, which has brought together public and private sector COVID-19 therapeutic and vaccine efforts, is intensely pursuing the development and testing of therapeutic monoclonal antibodies for COVID-19 [4]. Stay tuned for more information about these potentially significant advances in the next few months.
References:
[1] Humoral immune response and prolonged PCR positivity in a cohort of 1343 SARS-CoV 2 patients in the New York City region. Wajnberg A , Mansour M, Leven E, Bouvier NM, Patel G, Firpo A, Mendu R, Jhang J, Arinsburg S, Gitman M, Houldsworth J, Baine I, Simon V, Aberg J, Krammer F, Reich D, Cordon-Cardo C. medRxiv. Preprint Posted May 5, 2020.
[2] A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Wu Y. et al., Science. 13 May 2020 [Epub ahead of publication]
[3] Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Pinto D, Park YJ, Beltramello M, Veesler D, Cortil D, et al. Nature. 18 May 2020 [Epub ahead of print]
[4] Accelerating COVID-19 therapeutic interventions and vaccines (ACTIV): An unprecedented partnership for unprecedented times. Collins FS, Stoffels P. JAMA. 2020 May 18.
Links:
Coronavirus (COVID-19) (NIH)
Monoclonal Antibodies (National Cancer Institute/NIH)
Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV)
NIH Support: National Institute of Allergy and Infectious Diseases; National Institute of General Medical Sciences
Early Riser or Night Owl? New Study May Help to Explain the Difference
Posted on by Dr. Francis Collins

Credit: Clarisse Ricci, University of California, San Diego
Some people are early risers, wide awake at the crack of dawn. Others are night owls who can’t seem to get to bed until well after midnight and prefer to sleep in. Why is this? An NIH-funded team has some new clues based on evidence showing how a molecular “switch” wired into the biological clocks of extreme early risers leads them to operate on a daily cycle of about 20 hours instead of a full 24-hour, or circadian (Latin for “about a day”), cycle [1].
These new atomic-level details, shared from fruit flies to humans, may help to explain how more subtle clock variations predispose people to follow different sleep patterns. They also may lead to new treatments designed to reset the clock in people struggling with sleep disorders, jet lag, or night-shift work.
This work, published recently in the journal eLIFE, comes from Carrie Partch, University of California, Santa Cruz, and her colleagues at Duke-NUS Medical School in Singapore and the University of California, San Diego. It builds on decades of research into biological clocks, which help to control sleeping and waking, rest and activity, fluid balance, body temperature, cardiac rate, oxygen consumption, and even the secretions of endocrine glands.
These clocks, found in cells and tissues throughout the body, are composed of specialized sets of proteins. They interact in specific ways to regulate transcription of about 15 percent of the genome over a 24-hour period. All this interaction helps to align waking hours and other aspects of our physiology to the 24-hour passage of day and night.
In the latest paper, Partch and her colleagues focused on two core clock components: an enzyme known as casein kinase 1 (CK1) and a protein called PERIOD. Clock-altering mutations in CK1 and PERIOD have been known for many years. In fact, CK1 was discovered in studies of golden hamsters more than 20 years ago after researchers noticed one hamster that routinely woke up much earlier than the others [2,3].
It turns out that the timing of biological clocks is strongly influenced by the rise and fall of the PERIOD protein. This daily oscillation normally takes place over 24 hours, but that’s where CK1 enters the picture. The enzyme adjusts PERIOD levels by chemically modifying the protein at one of two sites, thereby adjusting its stability. When one site is modified, it keeps the protein protected and stable. At the other site, it leaves it unprotected and degradable.
Many of these details had been worked out over the years. But, Partch wanted to drill even deeper to answer an essential question: Why does this process normally take 24 hours, which is remarkably slow biochemically? And, what changes in those whose daily cycle gets cut far short?
To find out, her team performed a series of protein structure and biochemical analyses of the CK1 mutation originally found in hamsters, along with several other clock-altering versions of the enzyme found in organisms ranging from flies to humans. What they’ve discovered is a portion of CK1 acts as a switch. When this switch functions normally, it generates a near-perfect 24-hour cycle by keeping PERIOD’s stability just right. In this case, people easily and correctly align their internal clocks to the daily coming and going of daylight.
If the switch favors a faster breakdown of the protein, the daily cycle grows shorter and less tightly bound to daylight. For these early risers, it’s a constant struggle to adjust to life in a 24-hour world. Though they try to get in sync, these early risers are never able to catch up. Conversely, a switch that favors a slower breakdown will lengthen the clock, predisposing some to be night owls.
Such shifts in clock timing can arise from alterations either to the CK1 enzyme or the PERIOD protein. In fact, people with an inherited sleep disorder called Familial Advanced Sleep Phase Syndrome carry a mutation in the PERIOD protein at one of the places that CK1 modifies. The new work shows that this change makes PERIOD more stable by interfering with the enzyme’s ability to mark the protein for degradation.
One thing that makes the CK1 enzyme so fascinating is that it’s extremely ancient. A nearly identical version of the enzyme to the one in humans and hamsters can be found in single-celled green algae! It’s clear that this enzyme and its function in biological clocks is, evolutionarily speaking, rather special. And at one level, that makes total sense—our planet has operated on a 24-hour clock for the entire span of evolutionary time.
The versions of CK1 that Partch’s team studied here are rare in people. She now plans to study other variations that turn up in humans much more often.
Her discoveries are sure to offer a fascinating view on these internal clocks and, pardon the pun, how they make us all tick. She hopes they’ll lead to new ways to adjust the clock in those with sleep disorders and even the means to reset the clock in people who regularly travel overseas or work the night shift.
Ultimately, Partch would like to tap into the crosstalk between biological clocks and the ability of cells to repair their DNA. She wants to see if clock disruptions have any implications for cancer susceptibility. And yes, now’s a good time to find out the answer.
References:
[1] Casein kinase 1 dynamics underlie substrate selectivity and the PER2 circadian phosphoswitch. Philpott JM, Narasimamurthy R, Ricci CG, Freeberg AM, Hunt SR, Yee LE, Pelofsky RS, Tripathi S, Virshup DM, Partch CL. eLIFE. 2020 Feb 11;9.
[2] A mutation of the circadian system in golden hamsters. Ralph MR, Menaker M. Science. 1988 Sep 2;241(4870):1225-7.
[3] Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, Takahashi JS. Science. 2000 Apr 21;288(5465):483-92.
Links:
Circadian Rhythms (National Institute of General Medical Sciences/NIH)
Advanced Sleep Phase Syndrome, Familial (Genetic and Rare Disease Center/NIH)
Partch Lab (University of California, Santa Cruz)
NIH Support: National Institute of General Medical Sciences; Office of the Director
Deciphering Another Secret of Life
Posted on by Dr. Francis Collins

In 1953, Francis Crick famously told the surprised customers at the Eagle and Child pub in London that he and Jim Watson had discovered the secret of life. When NIH’s Marshall Nirenberg and his colleagues cracked the genetic code in 1961, it was called the solution to life’s greatest secret. Similarly, when the complete human genome sequence was revealed for the first time in 2003, commentators (including me) referred to this as the moment where the book of life for humans was revealed. But there are many more secrets of life that still need to be unlocked, including figuring out the biochemical rules of a protein shape-shifting phenomenon called allostery [1].
Among those taking on this ambitious challenge is a recipient of a 2018 NIH Director’s New Innovator Award, Srivatsan Raman of the University of Wisconsin-Madison. If successful, such efforts could revolutionize biology by helping us better understand how allosteric proteins reconfigure themselves in the right shapes at the right times to regulate cell signaling, metabolism, and many other important biological processes.
What exactly is an allosteric protein? Proteins have active, or orthosteric, sites that turn the proteins off or on when specific molecules bind to them. Some proteins also have less obvious regulatory, or allosteric, sites that indirectly affect the proteins’ activity when outside molecules bind to them. In many instances, allosteric binding triggers a change in the shape of the protein.
Allosteric proteins include oxygen-carrying hemoglobin and a variety of enzymes crucial to human health and development. In his work, Raman will start by studying a relatively simple bacterial protein, consisting of less than 200 amino acids, to understand the basics of how allostery works over time and space.
Raman, who is a synthetic biologist, got the idea for this project a few years ago while tinkering in the lab to modify an allosteric protein to bind new molecules. As part of the process, he and his team used a new technology called deep mutational scanning to study the functional consequences of removing individual amino acids from the protein [2].
The screen took them on a wild ride of unexpected functional changes, and a new research opportunity called out to him. He could combine this scanning technology with artificial intelligence and other cutting-edge imaging and computational tools to probe allosteric proteins more systematically in hopes of deciphering the basic molecular rules of allostery.
With the New Innovator Award, Raman’s group will first create a vast number of protein mutants to learn how best to determine the allosteric signaling pathway(s) within a protein. They want to dissect out the properties of each amino acid and determine which connect into a binding site and precisely how those linkages are formed. The researchers also want to know how the amino acids tend to configure into an inactive state and how that structure changes into an active state.
Based on these initial studies, the researchers will take the next step and use their dataset to predict where allosteric pathways are found in individual proteins. They will also try to figure out if allosteric signals are sent in one direction only or whether they can be bidirectional.
The experiments will be challenging, but Raman is confident that they will serve to build a more unified view of how allostery works. In fact, he hopes the data generated—and there will be a massive amount—will reveal novel sites to control or exploit allosteric signaling. Such information will not only expand fundamental biological understanding, but will accelerate efforts to discover new therapies for diseases, such as cancer, in which disruption of allosteric proteins plays a crucial role.
References:
[1] Allostery: an illustrated definition for the ‘second secret of life.’ Fenton AW. Trends Biochem Sci. 2008 Sep;33(9):420-425.
[2] Engineering an allosteric transcription factor to respond to new ligands. Taylor ND, Garruss AS, Moretti R, Chan S, Arbing MA, Cascio D, Rogers JK, Isaacs FJ, Kosuri S, Baker D, Fields S, Church GM, Raman S. Nat Methods. 2016 Feb;13(2):177-183.
Links:
Drug hunters explore allostery’s advantages. Jarvis LM, Chemical & Engineering News. 2019 March 10
Allostery: An Overview of Its History, Concepts, Methods, and Applications. Liu J, Nussinov R. PLoS Comput Biol. 2016 Jun 2;12(6):e1004966.
Srivatsan Raman (University of Wisconsin-Madison)
Raman Project Information (NIH RePORTER)
NIH Director’s New Innovator Award (Common Fund/NIH)
NIH Support: National Institute of General Medical Sciences; Common Fund
Tracking Peptides in Cell Soup
Posted on by Dr. Francis Collins

Credit: William Wimley, Tulane University, New Orleans
If you think this soup looks unappealing for this year’s Thanksgiving feast, you’re right! If you were crazy enough to take a sip, you’d find it to be virtually flavorless—just a salty base (red) with greasy lipid globules (green) floating on top. But what this colorful concoction lacks in taste, it makes up for as a valuable screening tool for peptides, miniature versions of proteins that our bodies use to control many cellular processes.
In this image, William Wimley, an NIH-supported researcher at Tulane University, New Orleans, has stirred up the soup and will soon add some peptides. These peptides aren’t made by our cells, though. They’re synthesized in the lab, allowing Wimley and team to tweak their chemical structures and hopefully create ones with therapeutic potential, particularly as smart-delivery systems to target cells with greater precision and deliver biological cargoes such as drugs [1].
Cryo-EM Images Capture Key Enzyme Tied to Cancer, Aging
Posted on by Dr. Francis Collins
Each time your cells divide, telomeres—complexes of specialized DNA sequences, RNA, and protein that protect the tips of your chromosomes—shorten just a bit. And, as the video shows, that shortening renders the genomic information on your chromosomes more vulnerable to changes that can drive cancer and other diseases of aging.
Consequently, over the last few decades, much research has focused on efforts to understand telomerase, a naturally occurring enzyme that helps to replace the bits of telomere lost during cell division. But there’s been a major hitch: until recently, scientists hadn’t been able to determine telomerase’s molecular structure in detail—a key step in figuring out exactly how the enzyme works. Now, thanks to better purification methods and an exciting technology called cryo-electron microscopy (cryo-EM), NIH-funded researchers and their colleagues have risen to the challenge to produce the most detailed view yet of human telomerase in its active form [1].
This structural biology advance is a critical step toward learning more about the role of telomerase in cancers, as well as genetic conditions linked to telomerase deficiencies. It’s also an important milestone in the quest for drugs targeting telomerase in different ways, perhaps to slow the growth of cancerous cells or to boost the proliferative capacity of life-giving adult stem cells.
One reason telomerase has been so difficult to study in humans is that the enzyme isn’t produced at detectable levels in the vast majority of our cells. To get around this problem, the team led by Eva Nogales and Kathleen Collins at the University of California, Berkeley, first coaxed human cells in the lab to produce larger quantities of active telomerase. They then used fluorescent microscopy, along with extensive knowledge of the enzyme’s biochemistry, to develop a multi-step purification process that yielded relatively homogenous samples of active telomerase.
The new study is also yet another remarkable example of how cryo-EM microscopy has opened up new realms of scientific possibility. That’s because, in comparison to other methods, cryo-EM enables researchers to solve complex macromolecular structures even when only tiny amounts of material are available. It can also produce detailed images of molecules, like telomerase, that are extremely flexible and hard to keep still while taking a picture of their structure.
As described in Nature, the researchers used cryo-EM to capture the structure of human telomerase in unprecedented detail. Their images reveal two lobes, held together by a flexible RNA tether. One of those lobes contains the highly specialized core enzyme. It uses an internal RNA template as a guide to make the repetitive, telomeric DNA that’s added at the tips of chromosomes. The second lobe, consisting of a complex of RNA and RNA-binding proteins, plays important roles in keeping the complex stable and properly in place.
This new, more-detailed view helps to explain how mutations in particular genes may lead to telomerase-related health conditions, including bone marrow failure, as well as certain forms of anemia and pulmonary fibrosis. For example, it reveals that a genetic defect known to cause bone marrow failure affects an essential protein in a spot that’s especially critical for telomerase’s proper conformation and function.
This advance will also be a big help for designing therapies that encourage telomerase activity. For example, it could help to boost the success of bone marrow transplants by rejuvenating adult stem cells. It might also be possible to reinforce the immune systems of people with HIV infections. While telomerase-targeted treatments surely won’t stop people from growing old, new insights into this important enzyme will help to understand aging better, including why some people appear to age faster than others.
As remarkable as these new images are, the researchers aren’t yet satisfied. They’ll continue to refine them down to the minutest structural details. They say they’d also like to use cryo-EM to understand better how the complex attaches to chromosomes to extend telomeres. Each new advance in the level of atomic detail will not only make for amazing new videos, it will help to advance understanding of human biology in health, aging, and disease.
References:
[1] Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nguyen THD, Tam J, Wu RA, Greber BJ, Toso D, Nogales E, Collins K. Nature. 2018 April 25. [Epub ahead of publication]
Links:
High Resolution Electron Microscopy (National Cancer Institute/NIH)
Nogales Lab (University of California, Berkeley)
Collins Lab (University of California, Berkeley)
NIH Support: National Institute of General Medical Sciences
Creative Minds: The Human Gut Microbiome’s Top 100 Hits
Posted on by Dr. Francis Collins

Michael Fishbach
Microbes that live in dirt often engage in their own deadly turf wars, producing a toxic mix of chemical compounds (also called “small molecules”) that can be a source of new antibiotics. When he started out in science more than a decade ago, Michael Fischbach studied these soil-dwelling microbes to look for genes involved in making these compounds.
Eventually, Fischbach, who is now at the University of California, San Francisco, came to a career-altering realization: maybe he didn’t need to dig in dirt! He hypothesized an even better way to improve human health might be found in the genes of the trillions of microorganisms that dwell in and on our bodies, known collectively as the human microbiome.
Muscle Enzyme Explains Weight Gain in Middle Age
Posted on by Dr. Francis Collins

Thinkstock/tetmc
The struggle to maintain a healthy weight is a lifelong challenge for many of us. In fact, the average American packs on an extra 30 pounds from early adulthood to age 50. What’s responsible for this tendency toward middle-age spread? For most of us, too many calories and too little exercise definitely play a role. But now comes word that another reason may lie in a strong—and previously unknown—biochemical mechanism related to the normal aging process.
An NIH-led team recently discovered that the normal process of aging causes levels of an enzyme called DNA-PK to rise in animals as they approach middle age. While the enzyme is known for its role in DNA repair, their studies show it also slows down metabolism, making it more difficult to burn fat. To see if reducing DNA-PK levels might rev up the metabolism, the researchers turned to middle-aged mice. They found that a drug-like compound that blocked DNA-PK activity cut weight gain in the mice by a whopping 40 percent!