mouse study
An Inflammatory View of Early Alzheimer’s Disease
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Detecting the earliest signs of Alzheimer’s disease (AD) in middle-aged people and tracking its progression over time in research studies continue to be challenging. But it is easier to do in shorter-lived mammalian models of AD, especially when paired with cutting-edge imaging tools that look across different regions of the brain. These tools can help basic researchers detect telltale early changes that might point the way to better prevention or treatment strategies in humans.
That’s the case in this technicolor snapshot showing early patterns of inflammation in the brain of a relatively young mouse bred to develop a condition similar to AD. You can see abnormally high levels of inflammation throughout the front part of the brain (orange, green) as well as in its middle part—the septum that divides the brain’s two sides. This level of inflammation suggests that the brain has been injured.
What’s striking is that no inflammation is detectable in parts of the brain rich in cholinergic neurons (pink), a distinct type of nerve cell that helps to control memory, movement, and attention. Though these neurons still remain healthy, researchers would like to know if the inflammation also will destroy them as AD progresses.
This colorful image comes from medical student Sakar Budhathoki, who earlier worked in the NIH labs of Lorna Role and David Talmage, National Institute of Neurological Disorders and Stroke (NINDS). Budhathoki, teaming with postdoctoral scientist Mala Ananth, used a specially designed wide-field scanner that sweeps across brain tissue to light up fluorescent markers and capture the image. It’s one of the scanning approaches pioneered in the Role and Talmage labs [1,2].
The two NIH labs are exploring possible links between abnormal inflammation and damage to the brain’s cholinergic signaling system. In fact, medications that target cholinergic function remain the first line of treatment for people with AD and other dementias. And yet, researchers still haven’t adequately determined when, why, and how the loss of these cholinergic neurons relates to AD.
It’s a rich area of basic research that offers hope for greater understanding of AD in the future. It’s also the source of some fascinating images like this one, which was part of the 2022 Show Us Your BRAIN! Photo and Video Contest, supported by NIH’s Brain Research Through Advancing Innovative Neurotechnologies® (BRAIN) Initiative.
References:
[1] NeuRegenerate: A framework for visualizing neurodegeneration. Boorboor S, Mathew S, Ananth M, Talmage D, Role LW, Kaufman AE. IEEE Trans Vis Comput Graph. 2021;Nov 10;PP.
[2] NeuroConstruct: 3D reconstruction and visualization of neurites in optical microscopy brain images. Ghahremani P, Boorboor S, Mirhosseini P, Gudisagar C, Ananth M, Talmage D, Role LW, Kaufman AE. IEEE Trans Vis Comput Graph. 2022 Dec;28(12):4951-4965.
Links:
Alzheimer’s Disease & Related Dementias (National Institute on Aging/NIH)
Role Lab (National Institute of Neurological Disorders and Stroke/NIH)
Talmage Lab (NINDS)
The Brain Research Through Advancing Innovative Neurotechnologies® (BRAIN) Initiative (NIH)
Show Us Your BRAINs! Photo and Video Contest (BRAIN Initiative)
NIH Support: National Institute of Neurological Disorders and Stroke
How the Brain Differentiates the ‘Click,’ ‘Crack,’ or ‘Thud’ of Everyday Tasks
Posted on by Lawrence Tabak, D.D.S., Ph.D.

If you’ve been staying up late to watch the World Series, you probably spent those nine innings hoping for superstars Bryce Harper or José Altuve to square up a fastball and send it sailing out of the yard. Long-time baseball fans like me can distinguish immediately the loud crack of a home-run swing from the dull thud of a weak grounder.
Our brains have such a fascinating ability to discern “right” sounds from “wrong” ones in just an instant. This applies not only in baseball, but in the things that we do throughout the day, whether it’s hitting the right note on a musical instrument or pushing the car door just enough to click it shut without slamming.
Now, an NIH-funded team of neuroscientists has discovered what happens in the brain when one hears an expected or “right” sound versus a “wrong” one after completing a task. It turns out that the mammalian brain is remarkably good at predicting both when a sound should happen and what it ideally ought to sound like. Any notable mismatch between that expectation and the feedback, and the hearing center of the brain reacts.
It may seem intuitive that humans and other animals have this auditory ability, but researchers didn’t know how neurons in the brain’s auditory cortex, where sound is processed, make these snap judgements to learn complex tasks. In the study published in the journal Current Biology, David Schneider, New York University, New York, set out to understand how this familiar experience really works.
To do it, Schneider and colleagues, including postdoctoral fellow Nicholas Audette, looked to mice. They are a lot easier to study in the lab than humans and, while their brains aren’t miniature versions of our own, our sensory systems share many fundamental similarities because we are both mammals.
Of course, mice don’t go around hitting home runs or opening and closing doors. So, the researchers’ first step was training the animals to complete a task akin to closing the car door. To do it, they trained the animals to push a lever with their paws in just the right way to receive a reward. They also played a distinctive tone each time the lever reached that perfect position.
After making thousands of attempts and hearing the associated sound, the mice knew just what to do—and what it should sound like when they did it right. Their studies showed that, when the researchers removed the sound, played the wrong sound, or played the correct sound at the wrong time, the mice took notice and adjusted their actions, just as you might do if you pushed a car door shut and the resulting click wasn’t right.
To find out how neurons in the auditory cortex responded to produce the observed behaviors, Schneider’s team also recorded brain activity. Intriguingly, they found that auditory neurons hardly responded when a mouse pushed the lever and heard the sound they’d learned to expect. It was only when something about the sound was “off” that their auditory neurons suddenly crackled with activity.
As the researchers explained, it seems from these studies that the mammalian auditory cortex responds not to the sounds themselves but to how those sounds match up to, or violate, expectations. When the researchers canceled the sound altogether, as might happen if you didn’t push a car door hard enough to produce the familiar click shut, activity within a select group of auditory neurons spiked right as they should have heard the sound.
Schneider’s team notes that the same brain areas and circuitry that predict and process self-generated sounds in everyday tasks also play a role in conditions such as schizophrenia, in which people may hear voices or other sounds that aren’t there. The team hopes their studies will help to explain what goes wrong—and perhaps how to help—in schizophrenia and other neural disorders. Perhaps they’ll also learn more about what goes through the healthy brain when anticipating the satisfying click of a closed door or the loud crack of a World Series home run.
Reference:
[1] Precise movement-based predictions in the mouse auditory cortex. Audette NJ, Zhou WX, Chioma A, Schneider DM. Curr Biology. 2022 Oct 24.
Links:
How Do We Hear? (National Institute on Deafness and Other Communication Disorders/NIH)
Schizophrenia (National Institute of Mental Health/NIH)
David Schneider (New York University, New York)
NIH Support: National Institute of Mental Health; National Institute on Deafness and Other Communication Disorders
The Amazing Brain: Seeing Two Memories at Once
Posted on by Lawrence Tabak, D.D.S., Ph.D.

The NIH’s Brain Research Through Advancing Innovative Neurotechnologies® (BRAIN) Initiative is revolutionizing our understanding of the human brain. As described in the initiative’s name, the development of innovative imaging technologies will enable researchers to see the brain in new and increasingly dynamic ways. Each year, the initiative celebrates some standout and especially creative examples of such advances in the “Show Us Your BRAINs! Photo & Video Contest. During most of August, I’ll share some of the most eye-catching developments in our blog series, The Amazing Brain.
In this fascinating image, you’re seeing two stored memories, which scientists call engrams, in the hippocampus region of a mouse’s brain. The engrams show the neural intersection of a good memory (green) and a bad memory (pink). You can also see the nuclei of many neurons (blue), including nearby neurons not involved in the memory formation.
This award-winning image was produced by Stephanie Grella in the lab of NIH-supported neuroscientist Steve Ramirez, Boston University, MA. It’s also not the first time that the blog has featured Grella’s technical artistry. Grella, who will soon launch her own lab at Loyola University, Chicago, previously captured what a single memory looks like.
To capture two memories at once, Grella relied on a technology known as optogenetics. This powerful method allows researchers to genetically engineer neurons and selectively activate them in laboratory mice using blue light. In this case, Grella used a harmless virus to label neurons involved in recording a positive experience with a light-sensitive molecule, known as an opsin. Another molecular label was used to make those same cells appear green when activated.
After any new memory is formed, there’s a period of up to about 24 hours during which the memory is malleable. Then, the memory tends to stabilize. But with each retrieval, the memory can be modified as it restabilizes, a process known as memory reconsolidation.
Grella and team decided to try to use memory reconsolidation to their advantage to neutralize an existing fear. To do this, they placed their mice in an environment that had previously startled them. When a mouse was retrieving a fearful memory (pink), the researchers activated with light associated with the positive memory (green), which for these particular mice consisted of positive interactions with other mice. The aim was to override or disrupt the fearful memory.
As shown by the green all throughout the image, the experiment worked. While the mice still showed some traces of the fearful memory (pink), Grella explained that the specific cells that were the focus of her study shifted to the positive memory (green).
What’s perhaps even more telling is that the evidence suggests the mice didn’t just trade one memory for another. Rather, it appears that activating a positive memory actually suppressed or neutralized the animal’s fearful memory. The hope is that this approach might one day inspire methods to help people overcome negative and unwanted memories, such as those that play a role in post-traumatic stress disorder (PTSD) and other mental health issues.
Links:
Stephanie Grella (Boston University, MA)
Ramirez Group (Boston University)
Brain Research through Advancing Innovative Neurotechnologies® (BRAIN) Initiative (NIH)
Show Us Your BRAINs Photo & Video Contest (BRAIN Initiative)
NIH Support: BRAIN Initiative; Common Fund
‘Decoy’ Protein Works Against Multiple Coronavirus Variants in Early Study
Posted on by Lawrence Tabak, D.D.S., Ph.D.

The NIH continues to support the development of some very innovative therapies to control SARS-CoV-2, the coronavirus that causes COVID-19. One innovative idea involves a molecular decoy to thwart the coronavirus.
How’s that? The decoy is a specially engineered protein particle that mimics the 3D structure of the ACE2 receptor, a protein on the surface of our cells that the virus’s spike proteins bind to as the first step in causing an infection.
The idea is when these ACE2 decoys are administered therapeutically, they will stick to the spike proteins that crown the coronavirus (see image above). With its spikes covered tightly in decoy, SARS-CoV-2 has a more-limited ability to attach to the real ACE2 and infect our cells.
Recently, the researchers published their initial results in the journal Nature Chemical Biology, and the early data look promising [1]. They found in mouse models of severe COVID-19 that intravenous infusion of an engineered ACE2 decoy prevented lung damage and death. Though more study is needed, the researchers say the decoy therapy could potentially be delivered directly to the lungs through an inhaler and used alone or in combination with other COVID-19 treatments.
The findings come from a research team at the University of Illinois Chicago team, led by Asrar Malik and Jalees Rehman, working in close collaboration with their colleagues at the University of Illinois Urbana-Champaign. The researchers had been intrigued by an earlier clinical trial testing the ACE2 decoy strategy [2]. However, in this earlier attempt, the clinical trial found no reduction in mortality. The ACE2 drug candidate, which is soluble and degrades in the body, also proved ineffective in neutralizing the virus.
Rather than give up on the idea, the UIC team decided to give it a try. They engineered a new soluble version of ACE2 that structurally might work better as a decoy than the original one. Their version of ACE2, which includes three changes in the protein’s amino acid building blocks, binds the SARS-CoV-2 spike protein much more tightly. In the lab, it also appeared to neutralize the virus as well as monoclonal antibodies used to treat COVID-19.
To put it to the test, they conducted studies in mice. Normal mice don’t get sick from SARS-CoV-2 because the viral spike can’t bind well to the mouse version of the ACE2 receptor. So, the researchers did their studies in a mouse that carries the human ACE2 and develops a severe acute respiratory syndrome somewhat similar to that seen in humans with severe COVID-19.
In their studies, using both the original viral isolate from Washington State and the Gamma variant (P.1) first detected in Brazil, they found that infected mice infused with their therapeutic ACE2 protein had much lower mortality and showed few signs of severe acute respiratory syndrome. While the protein worked against both versions of the virus, infection with the more aggressive Gamma variant required earlier treatment. The treated mice also regained their appetite and weight, suggesting that they were making a recovery.
Further studies showed that the decoy bound to spike proteins from every variant tested, including Alpha, Beta, Delta and Epsilon. (Omicron wasn’t yet available at the time of the study.) In fact, the decoy bound just as well, if not better, to new variants compared to the original virus.
The researchers will continue their preclinical work. If all goes well, they hope to move their ACE2 decoy into a clinical trial. What’s especially promising about this approach is it could be used in combination with treatments that work in other ways, such as by preventing virus that’s already infected cells from growing or limiting an excessive and damaging immune response to the infection.
Last week, more than 17,500 people in the United States were hospitalized with severe COVID-19. We’ve got to continue to do all we can to save lives, and it will take lots of innovative ideas, like this ACE2 decoy, to put us in a better position to beat this virus once and for all.
References:
[1] Engineered ACE2 decoy mitigates lung injury and death induced by SARS-CoV-2 variants.
Zhang L, Dutta S, Xiong S, Chan M, Chan KK, Fan TM, Bailey KL, Lindeblad M, Cooper LM, Rong L, Gugliuzza AF, Shukla D, Procko E, Rehman J, Malik AB. Nat Chem Biol. 2022 Jan 19.
[2] Recombinant human angiotensin-converting enzyme 2 (rhACE2) as a treatment for patients with COVID-19 (APN01-COVID-19). ClinicalTrials.gov.
Links:
COVID-19 Research (NIH)
Accelerating COVID-19 Therapeutic Interventions and Vaccines (NIH)
Asrar Malik (University of Illinois Chicago)
Jalees Rehman (University of Illinois Chicago)
NIH Support: National Heart, Lung, and Blood Institute; National Institute of Allergy and Infectious Diseases
Engineering a Better Way to Deliver Therapeutic Genes to Muscles
Posted on by Dr. Francis Collins

Amid all the progress toward ending the COVID-19 pandemic, it’s worth remembering that researchers here and around the world continue to make important advances in tackling many other serious health conditions. As an inspiring NIH-supported example, I’d like to share an advance on the use of gene therapy for treating genetic diseases that progressively degenerate muscle, such as Duchenne muscular dystrophy (DMD).
As published recently in the journal Cell, researchers have developed a promising approach to deliver therapeutic genes and gene editing tools to muscle more efficiently, thus requiring lower doses [1]. In animal studies, the new approach has targeted muscle far more effectively than existing strategies. It offers an exciting way forward to reduce unwanted side effects from off-target delivery, which has hampered the development of gene therapy for many conditions.
In boys born with DMD (it’s an X-linked disease and therefore affects males), skeletal and heart muscles progressively weaken due to mutations in a gene encoding a critical muscle protein called dystrophin. By age 10, most boys require a wheelchair. Sadly, their life expectancy remains less than 30 years.
The hope is gene therapies will one day treat or even cure DMD and allow people with the disease to live longer, high-quality lives. Unfortunately, the benign adeno-associated viruses (AAVs) traditionally used to deliver the healthy intact dystrophin gene into cells mostly end up in the liver—not in muscles. It’s also the case for gene therapy of many other muscle-wasting genetic diseases.
The heavy dose of viral vector to the liver is not without concern. Recently and tragically, there have been deaths in a high-dose AAV gene therapy trial for X-linked myotubular myopathy (XLMTM), a different disorder of skeletal muscle in which there may already be underlying liver disease, potentially increasing susceptibility to toxicity.
To correct this concerning routing error, researchers led by Mohammadsharif Tabebordbar in the lab of Pardis Sabeti, Broad Institute of MIT and Harvard and Harvard University, Cambridge, MA, have now assembled an optimized collection of AAVs. They have been refined to be about 10 times better at reaching muscle fibers than those now used in laboratory studies and clinical trials. In fact, researchers call them myotube AAVs, or MyoAAVs.
MyoAAVs can deliver therapeutic genes to muscle at much lower doses—up to 250 times lower than what’s needed with traditional AAVs. While this approach hasn’t yet been tried in people, animal studies show that MyoAAVs also largely avoid the liver, raising the prospect for more effective gene therapies without the risk of liver damage and other serious side effects.
In the Cell paper, the researchers demonstrate how they generated MyoAAVs, starting out with the commonly used AAV9. Their goal was to modify the outer protein shell, or capsid, to create an AAV that would be much better at specifically targeting muscle. To do so, they turned to their capsid engineering platform known as, appropriately enough, DELIVER. It’s short for Directed Evolution of AAV capsids Leveraging In Vivo Expression of transgene RNA.
Here’s how DELIVER works. The researchers generate millions of different AAV capsids by adding random strings of amino acids to the portion of the AAV9 capsid that binds to cells. They inject those modified AAVs into mice and then sequence the RNA from cells in muscle tissue throughout the body. The researchers want to identify AAVs that not only enter muscle cells but that also successfully deliver therapeutic genes into the nucleus to compensate for the damaged version of the gene.
This search delivered not just one AAV—it produced several related ones, all bearing a unique surface structure that enabled them specifically to target muscle cells. Then, in collaboration with Amy Wagers, Harvard University, Cambridge, MA, the team tested their MyoAAV toolset in animal studies.
The first cargo, however, wasn’t a gene. It was the gene-editing system CRISPR-Cas9. The team found the MyoAAVs correctly delivered the gene-editing system to muscle cells and also repaired dysfunctional copies of the dystrophin gene better than the CRISPR cargo carried by conventional AAVs. Importantly, the muscles of MyoAAV-treated animals also showed greater strength and function.
Next, the researchers teamed up with Alan Beggs, Boston Children’s Hospital, and found that MyoAAV was effective in treating mouse models of XLMTM. This is the very condition mentioned above, in which very high dose gene therapy with a current AAV vector has led to tragic outcomes. XLMTM mice normally die in 10 weeks. But, after receiving MyoAAV carrying a corrective gene, all six mice had a normal lifespan. By comparison, mice treated in the same way with traditional AAV lived only up to 21 weeks of age. What’s more, the researchers used MyoAAV at a dose 100 times lower than that currently used in clinical trials.
While further study is needed before this approach can be tested in people, MyoAAV was also used to successfully introduce therapeutic genes into human cells in the lab. This suggests that the early success in animals might hold up in people. The approach also has promise for developing AAVs with potential for targeting other organs, thereby possibly providing treatment for a wide range of genetic conditions.
The new findings are the result of a decade of work from Tabebordbar, the study’s first author. His tireless work is also personal. His father has a rare genetic muscle disease that has put him in a wheelchair. With this latest advance, the hope is that the next generation of promising gene therapies might soon make its way to the clinic to help Tabebordbar’s father and so many other people.
Reference:
[1] Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Tabebordbar M, Lagerborg KA, Stanton A, King EM, Ye S, Tellez L, Krunnfusz A, Tavakoli S, Widrick JJ, Messemer KA, Troiano EC, Moghadaszadeh B, Peacker BL, Leacock KA, Horwitz N, Beggs AH, Wagers AJ, Sabeti PC. Cell. 2021 Sep 4:S0092-8674(21)01002-3.
Links:
Muscular Dystrophy Information Page (National Institute of Neurological Disorders and Stroke/NIH)
X-linked myotubular myopathy (Genetic and Rare Diseases Information Center/National Center for Advancing Translational Sciences/NIH)
Somatic Cell Genome Editing (Common Fund/NIH)
Mohammadsharif Tabebordbar (Broad Institute of MIT and Harvard and Harvard University, Cambridge, MA)
Sabeti Lab (Broad Institute of MIT and Harvard and Harvard University)
NIH Support: Eunice Kennedy Shriver National Institute of Child Health and Human Development; Common Fund
CRISPR-Based Anti-Viral Therapy Could One Day Foil the Flu—and COVID-19
Posted on by Dr. Francis Collins

CRISPR gene-editing technology has tremendous potential for making non-heritable DNA changes that can treat or even cure a wide range of devastating disorders, from HIV to muscular dystrophy Now, a recent animal study shows that another CRISPR system—targeting viral RNA instead of human DNA—could work as an inhaled anti-viral therapeutic that can be preprogrammed to seek out and foil potentially almost any flu strain and many other respiratory viruses, including SARS-CoV-2, the coronavirus that causes COVID-19.
How can that be? Other CRISPR gene-editing systems rely on a sequence-specific guide RNA to direct a scissor-like, bacterial enzyme (Cas9) to just the right spot in the genome to cut out, replace, or repair disease-causing mutations. This new anti-viral CRISPR system also relies on guide RNA. But the guide instead directs a different bacterial enzyme, called Cas13a, to the right spot in the viral genome to bind and cleave viral RNA and stop viruses from replicating in lung cells.
The findings, recently published in the journal Nature Biotechnology [1], come from the lab of Philip Santangelo, Georgia Institute of Technology and Emory University, Atlanta. Earlier studies by other groups had shown the potential of Cas13 for degrading the RNA of influenza viruses in a lab dish [2,3]. In this latest work, Santangelo and colleagues turned to mice and hamsters to see whether this enzyme could actually work in the lung tissue of a living animal.
What’s interesting is how Santangelo’s team did it. Rather than delivering the Cas13a protein itself to the lungs, the CRISPR system works by supplying a messenger RNA (mRNA) with the instructions to make the anti-viral Cas13a protein. This is the same idea as the Pfizer and Moderna mRNA-based COVID-19 vaccines, which temporarily direct your muscle cells to produce viral spike proteins that launch an immune response. In this case, the lung cells translate the Cas13a mRNA to produce the protein. Directed by the guide RNA that was also delivered to the same cells, Cas13a degrades the viral RNA and stops the infection. Because mRNA doesn’t enter the cell’s nucleus, it doesn’t interact with DNA and raise potential concerns about causing unwanted genetic changes.
The researchers designed guide RNAs that were specific to a shared, highly conserved portion of influenza viruses involved in replicating their genome and infecting other cells. They also designed another set directed to key portions of SARS-CoV-2.
Next, they delivered the Cas13a mRNA and guides straight to the lungs of animals using an adapted nebulizer, just like those used to deliver medicines to the lungs of people. In mice with influenza, Cas13a degraded influenza RNA in the lungs and the animals recovered without any apparent side effects. In SARS-CoV-2-infected hamsters, the same approach limited the virus’s ability to replicate in cells as the animals COVID-19-like symptoms improved.
The findings are the first to show that mRNA can be used to express the Cas13a protein in living lung tissue, not just in cells in a dish. It’s also the first to show that the bacterial Cas13a protein is effective at slowing or stopping replication of SARS-CoV-2. The latter raises hope that this CRISPR system could be quickly adapted to fight any future novel coronaviruses that develop the ability to infect humans.
The researchers report that this approach has potential to work against the vast majority—99 percent—of the flu strains that have circulated around the world over the last century. It also should be equally effective against the new and more contagious variants of SARS-CoV-2 now circulating around the globe. While more study is needed to understand the safety of such an anti-viral approach before trying it in humans, what’s clear is basic research advances like this one hold great potential for helping us to fight life-threatening respiratory viruses of the past, present, and future.
References:
[1] Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Blanchard EL, Vanover D, Bawage SS, Tiwari PM, Rotolo L, Beyersdorf J, Peck HE, Bruno NC, Hincapie R, Michel F, Murray J, Sadhwani H, Vanderheyden B, Finn MG, Brinton MA, Lafontaine ER, Hogan RJ, Zurla C, Santangelo PJ. Nat Biotechnol. 2021 Feb 3. [Published online ahead of print.]
[2] Programmable inhibition and detection of RNA viruses using Cas13. Freije CA, Myhrvold C, Boehm CK, Lin AE, Welch NL, Carter A, Metsky HC, Luo CY, Abudayyeh OO, Gootenberg JS, Yozwiak NL, Zhang F, Sabeti PC. Mol Cell. 2019 Dec 5;76(5):826-837.e11.
[3] Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. Abbott TR, Dhamdhere G, Liu Y, Lin X, Goudy L, Zeng L, Chemparathy A, Chmura S, Heaton NS, Debs R, Pande T, Endy D, La Russa MF, Lewis DB, Qi LS. Cell. 2020 May 14;181(4):865-876.e12.
Links:
COVID-19 Research (NIH)
Influenza (National Institute of Allergy and Infectious Diseases/NIH)
Santangelo Lab (Georgia Institute of Technology, Atlanta)
DNA Base Editing May Treat Progeria, Study in Mice Shows
Posted on by Dr. Francis Collins

My good friend Sam Berns was born with a rare genetic condition that causes rapid premature aging. Though Sam passed away in his teens from complications of this condition, called Hutchinson-Gilford progeria syndrome, he’s remembered today for his truly positive outlook on life. Sam expressed it, in part, by his willingness to make adjustments that allowed him, in his words, to put things that he always wanted to do in the “can do” category.
In this same spirit on behalf of the several hundred kids worldwide with progeria and their families, a research collaboration, including my NIH lab, has now achieved a key technical advance to move non-heritable gene editing another step closer to the “can do” category to treat progeria. As published in the journal Nature, our team took advantage of new gene-editing tools to correct for the first time a single genetic misspelling responsible for progeria in a mouse model, with dramatically beneficial effects [1, 2]. This work also has implications for correcting similar single-base typos that cause other inherited genetic disorders.
The outcome of this work is incredibly gratifying for me. In 2003, my NIH lab discovered the DNA mutation that causes progeria. One seemingly small glitch—swapping a “T” in place of a “C” in a gene called lamin A (LMNA)—leads to the production of a toxic protein now known as progerin. Without treatment, children with progeria develop normally intellectually but age at an exceedingly rapid pace, usually dying prematurely from heart attacks or strokes in their early teens.
The discovery raised the possibility that correcting this single-letter typo might one day help or even cure children with progeria. But back then, we lacked the needed tools to edit DNA safely and precisely. To be honest, I didn’t think that would be possible in my lifetime. Now, thanks to advances in basic genomic research, including work that led to the 2020 Nobel Prize in Chemistry, that’s changed. In fact, there’s been substantial progress toward using gene-editing technologies, such as the CRISPR editing system, for treating or even curing a wide range of devastating genetic conditions, such as sickle cell disease and muscular dystrophy
It turns out that the original CRISPR system, as powerful as it is, works better at knocking out genes than correcting them. That’s what makes some more recently developed DNA editing agents and approaches so important. One of them, which was developed by David R. Liu, Broad Institute of MIT and Harvard, Cambridge, MA, and his lab members, is key to these latest findings on progeria, reported by a team including my lab in NIH’s National Human Genome Research Institute and Jonathan Brown, Vanderbilt University Medical Center, Nashville, TN.
The relatively new gene-editing system moves beyond knock-outs to knock-ins [3,4]. Here’s how it works: Instead of cutting DNA as CRISPR does, base editors directly convert one DNA letter to another by enzymatically changing one DNA base to become a different base. The result is much like the find-and-replace function used to fix a typo in a word processor. What’s more, the gene editor does this without cutting the DNA.
Our three labs (Liu, Brown, and Collins) first teamed up with the Progeria Research Foundation, Peabody, MA, to obtain skin cells from kids with progeria. In lab studies, we found that base editors, targeted by an appropriate RNA guide, could successfully correct the LMNA gene in those connective tissue cells. The treatment converted the mutation back to the normal gene sequence in an impressive 90 percent of the cells.
But would it work in a living animal? To get the answer, we delivered a single injection of the DNA-editing apparatus into nearly a dozen mice either three or 14 days after birth, which corresponds in maturation level roughly to a 1-year-old or 5-year-old human. To ensure the findings in mice would be as relevant as possible to a future treatment for use in humans, we took advantage of a mouse model of progeria developed in my NIH lab in which the mice carry two copies of the human LMNA gene variant that causes the condition. Those mice develop nearly all of the features of the human illness
In the live mice, the base-editing treatment successfully edited in the gene’s healthy DNA sequence in 20 to 60 percent of cells across many organs. Many cell types maintained the corrected DNA sequence for at least six months—in fact, the most vulnerable cells in large arteries actually showed an almost 100 percent correction at 6 months, apparently because the corrected cells had compensated for the uncorrected cells that had died out. What’s more, the lifespan of the treated animals increased from seven to almost 18 months. In healthy mice, that’s approximately the beginning of old age.
This is the second notable advance in therapeutics for progeria in just three months. Last November, based on preclinical work from my lab and clinical trials conducted by the Progeria Research Foundation in Boston, the Food and Drug Administration (FDA) approved the first treatment for the condition. It is a drug called Zokinvy, and works by reducing the accumulation of progerin [5]. With long-term treatment, the drug is capable of extending the life of kids with progeria by 2.5 years and sometimes more. But it is not a cure.
We are hopeful this gene editing work might eventually lead to a cure for progeria. But mice certainly aren’t humans, and there are still important steps that need to be completed before such a gene-editing treatment could be tried safely in people. In the meantime, base editors and other gene editing approaches keep getting better—with potential application to thousands of genetic diseases where we know the exact gene misspelling. As we look ahead to 2021, the dream envisioned all those years ago about fixing the tiny DNA typo responsible for progeria is now within our grasp and getting closer to landing in the “can do” category.
References:
[1] In vivo base editing rescues Hutchinson-Gilford Progeria Syndrome in mice. Koblan LW et al. Nature. 2021 Jan 6.
[2] Base editor repairs mutation found in the premature-ageing syndrome progeria. Vermeij WP, Hoeijmakers JHJ. Nature. 6 Jan 2021.
[3] Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Nature. 2016 May 19;533(7603):420-424.
[4] Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Nature. 2017 Nov 23;551(7681):464-471.
[5] FDA approves first treatment for Hutchinson-Gilford progeria syndrome and some progeroid laminopathies. Food and Drug Administration. 2020 Nov 20.
Links:
Progeria (Genetic and Rare Diseases Information Center/NIH)
What are Genome Editing and CRISPR-Cas9? (National Library of Medicine/NIH)
Somatic Cell Genome Editing Program (Common Fund/NIH)
David R. Liu (Harvard University, Cambridge, MA)
Collins Group (National Human Genome Research Institute/NIH)
Jonathan Brown (Vanderbilt University Medical Center, Nashville, TN)
NIH Support: National Human Genome Research Institute; National Center for Advancing Translational Sciences; National Institute of Biomedical Imaging and Bioengineering; National Institute of Allergy and Infectious Diseases; National Institute of General Medical Sciences; Common Fund
Connecting Senescent Cells to Obesity and Anxiety
Posted on by Dr. Francis Collins
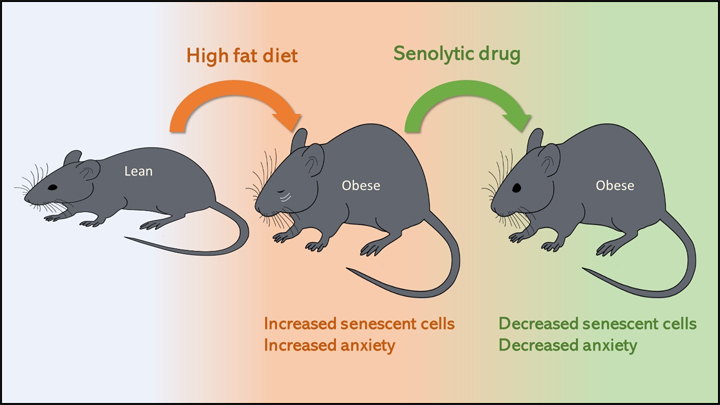
Obesity—which affects about 4 in 10 U.S. adults—increases the risk for lots of human health problems: diabetes, heart disease, certain cancers, and even anxiety and depression [1]. It’s also been associated with increased accumulation of senescent cells, which are older cells that resist death even as they lose the ability to grow and divide.
Now, NIH-funded researchers have found that when lean mice are fed a high-fat diet that makes them obese, they also have more senescent cells in their brain and show more anxious behaviors [2]. The researchers could reduce this obesity-driven anxiety using so-called senolytic drugs that cleared away the senescent cells. These findings are among the first to provide proof-of-concept that senolytics may offer a new avenue for treating an array of neuropsychiatric disorders, in addition to many other chronic conditions.
As we age, senescent cells accumulate in many parts of the body [3]. But cells can also enter a senescent state at any point in life in response to major stresses, such as DNA damage or chronic infection. Studies suggest that having lots of senescent cells around, especially later in life, is associated with a wide variety of chronic conditions, including osteoporosis, osteoarthritis, vascular disease, and general frailty.
Senescent cells display a “zombie”-like behavior known as a senescence-associated secretory phenotype (SASP). In this death-defying, zombie-like state, the cells ramp up their release of proteins, bioactive lipids, DNA, and other factors that, like a zombie virus, induce nearby healthy cells to join in the dysfunction.
In fact, the team behind this latest study, led by James Kirkland, Mayo Clinic, Rochester, MN, recently showed that transplanting small numbers of senescent cells into young mice is enough to cause them weakness, frailty, and persistent health problems. Those ill effects were alleviated with a senolytic cocktail, including dasatinib (a leukemia drug) and quercetin (a plant compound). This drug cocktail overrode the zombie-like SASP phenotype and forced the senescent cells to undergo programmed cell death and finally die.
Previous research indicates that senescent cells also accumulate in obesity, and not just in adipose tissues. Moreover, recent studies have linked senescent cells in the brain to neurodegenerative conditions, including Alzheimer’s disease, and showed in mice that dasatinib and quercetin helps to alleviate neurodegenerative disease [4,5]. In the latest paper, published in the journal Cell Metabolism, Kirkland and colleagues asked whether senescent cells in the brain also could explain anxiety-like behavior in obesity.
The answer appears to be “yes.” The researchers showed that lean mice, if allowed to feast on a high-fat diet, grew obese and became more anxious about exploring open spaces and elevated mazes.
The researchers also found that the obese mice had an increase in senescent cells in the white matter near the lateral ventricle, a part of the brain that offers a pathway for cerebrospinal fluid. Those senescent cells also contained an excessive amount of fat. Could senolytic drugs clear those cells and make the obesity-related anxiety go away?
To find out, the researchers treated lean and obese mice with a senolytic drug for 10 weeks. The treatment didn’t lead to any changes in body weight. But, as senescent cells were cleared from their brains, the obese mice showed a significant reduction in their anxiety-related behavior. They lost their anxiety without losing the weight!
More preclinical study is needed to understand more precisely how the treatment works. But, it’s worth noting that clinical trials testing a variety of senolytic drugs are already underway for many conditions associated with senescent cells, including chronic kidney disease [6,7], frailty [8], and premature aging associated with bone marrow transplant [9].
As a matter of fact, just after the Cell Metabolism paper came out, Kirkland’s team published encouraging though preliminary, first-in-human results of the previously mentioned senolytic drug dasatinib in 14 people with age-related idiopathic pulmonary fibrosis, a condition in which lung tissue becomes damaged and scarred [10]. Caution is warranted as we learn more about the associated risks and benefits, but it’s safe to say we’ll be hearing a lot more about senolytics in the years ahead.
References:
[1] Adult obesity facts (Centers for Disease Control and Prevention)
[2] Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Ogrodnik M et al. Cell Metabolism. 2019 Jan 3.
[3] Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. Tchkonia T, Kirkland JL. JAMA. 2018 Oct 2;320(13):1319-1320.
[4] Tau protein aggregation is associated with cellular senescence in the brain. Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, Orr ME. Aging Cell. 2018 Dec;17(6):e12840.
[5] Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Nature. 2018 Oct;562(7728):578-582.
[6] Inflammation and Stem Cells in Diabetic and Chronic Kidney Disease. ClinicalTrials.gov, Sep 2018.
[7] Senescence in Chronic Kidney Disease. Clinicaltrials.gov, Sep 2018.
[8] Alleviation by Fisetin of Frailty, Inflammation, and Related Measures in Older Adults (AFFIRM-LITE). Clinicaltrials.gov, Dec 2018.
[9] Hematopoietic Stem Cell Transplant Survivors Study (HTSS Study). Clinicaltrials.gov, Sep 2018.
[10] Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. Justice JN, Nambiar AN, Tchkonia T, LeBrasseur K, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, Kirkland JL. EBioMed. 5 Jan. 2019. [Epub ahead of print]
Links:
Healthy Aging (National Institute on Aging/NIH)
Video: Vail Scientific Summit James Kirkland Interview (Youtube)
James Kirkland (Mayo Clinic, Rochester, MN)
NIH Support: National Institute on Aging; National Institute of Neurological Disorders and Stroke